4:16 PM 7/29/2020
- Get link
- X
- Other Apps
All News Review In 25 Saved Stories

| Saved Stories - None | ||
|---|---|---|
| Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic | Nature Microbiology | ||
Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic
Abstract
There are outstanding evolutionary questions on the recent emergence of human coronavirus SARS-CoV-2 including the role of reservoir species, the role of recombination and its time of divergence from animal viruses. We find that the sarbecoviruses—the viral subgenus containing SARS-CoV and SARS-CoV-2—undergo frequent recombination and exhibit spatially structured genetic diversity on a regional scale in China. SARS-CoV-2 itself is not a recombinant of any sarbecoviruses detected to date, and its receptor-binding motif, important for specificity to human ACE2 receptors, appears to be an ancestral trait shared with bat viruses and not one acquired recently via recombination. To employ phylogenetic dating methods, recombinant regions of a 68-genome sarbecovirus alignment were removed with three independent methods. Bayesian evolutionary rate and divergence date estimates were shown to be consistent for these three approaches and for two different prior specifications of evolutionary rates based on HCoV-OC43 and MERS-CoV. Divergence dates between SARS-CoV-2 and the bat sarbecovirus reservoir were estimated as 1948 (95% highest posterior density (HPD): 1879–1999), 1969 (95% HPD: 1930–2000) and 1982 (95% HPD: 1948–2009), indicating that the lineage giving rise to SARS-CoV-2 has been circulating unnoticed in bats for decades.
Main
In December 2019, a cluster of pneumonia cases epidemiologically linked to an open-air live animal market in the city of Wuhan (Hubei Province), China1,2 led local health officials to issue an epidemiological alert to the Chinese Center for Disease Control and Prevention and the World Health Organization’s (WHO) China Country Office. In early January, the aetiological agent of the pneumonia cases was found to be a coronavirus3, subsequently named SARS-CoV-2 by an International Committee on Taxonomy of Viruses (ICTV) Study Group4 and also named hCoV-19 by Wu et al.5. The first available sequence data6 placed this novel human pathogen in the Sarbecovirus subgenus of Coronaviridae7, the same subgenus as the SARS virus that caused a global outbreak of >8,000 cases in 2002–2003. By mid-January 2020, the virus was spreading widely within Hubei province and by early March SARS-CoV-2 was declared a pandemic8.
In outbreaks of zoonotic pathogens, identification of the infection source is crucial because this may allow health authorities to separate human populations from the wildlife or domestic animal reservoirs posing the zoonotic risk9,10. If stopping an outbreak in its early stages is not possible—as was the case for the COVID-19 epidemic in Hubei—identification of origins and point sources is nevertheless important for containment purposes in other provinces and prevention of future outbreaks. When the first genome sequence of SARS-CoV-2, Wuhan-Hu-1, was released on 10 January 2020 (GMT) on Virological.org by a consortium led by Zhang6, it enabled immediate analyses of its ancestry. Across a large region of the virus genome, corresponding approximately to ORF1b, it did not cluster with any of the known bat coronaviruses indicating that recombination probably played a role in the evolutionary history of these viruses5,7. Subsequently a bat sarbecovirus—RaTG13, sampled from a Rhinolophus affinis horseshoe bat in 2013 in Yunnan Province—was reported that clusters with SARS-CoV-2 in almost all genomic regions with approximately 96% genome sequence identity2. Zhou et al.2 concluded from the genetic proximity of SARS-CoV-2 to RaTG13 that a bat origin for the current COVID-19 outbreak is probable. Concurrent evidence also proposed pangolins as a potential intermediate species for SARS-CoV-2 emergence and suggested them as a potential reservoir species11,12,13.
Unlike other viruses that have emerged in the past two decades, coronaviruses are highly recombinogenic14,15,16. Influenza viruses reassort17 but they do not undergo homologous recombination within RNA segments18,19, meaning that origins questions for influenza outbreaks can always be reduced to origins questions for each of influenza’s eight RNA segments. For coronaviruses, however, recombination means that small genomic subregions can have independent origins, identifiable if sufficient sampling has been done in the animal reservoirs that support the endemic circulation, co-infection and recombination that appear to be common. Here, we analyse the evolutionary history of SARS-CoV-2 using available genomic data on sarbecoviruses. We demonstrate that the sarbecoviruses circulating in horseshoe bats have complex recombination histories as reported by others15,20,21,22,23,24,25,26. Despite the SARS-CoV-2 lineage’s acquisition of residues in its Spike (S) protein’s receptor-binding domain (RBD) permitting the use of human ACE2 (ref. 27) receptors and its RBD being genetically closer to a pangolin virus than to RaTG13 (refs. 11,12,13,22,28)—a signal that suggests recombination—the divergence patterns in the S protein do not show evidence of recombination between the lineage leading to SARS-CoV-2 and known sarbecoviruses. Our results indicate the presence of a single lineage circulating in bats with properties that allowed it to infect human cells, as previously described for bat sarbecoviruses related to the first SARS-CoV lineage29,30,31.
To gauge the length of time this lineage has circulated in bats, we estimate the time to the most recent common ancestor (TMRCA) of SARS-CoV-2 and RaTG13. We use three bioinformatic approaches to remove the effects of recombination, and we combine these approaches to identify putative non-recombinant regions that can be used for reliable phylogenetic reconstruction and dating. Collectively our analyses point to bats being the primary reservoir for the SARS-CoV-2 lineage. While it is possible that pangolins, or another hitherto undiscovered species, may have acted as an intermediate host facilitating transmission to humans, current evidence is consistent with the virus having evolved in bats resulting in bat sarbecoviruses that can replicate in the upper respiratory tract of both humans and pangolins25,32.
ResultsRecombination analysis and identification of breakpoint-free genome regions
Among the 68 sequences in the aligned sarbecovirus sequence set, 67 show evidence of mosaicism (all Dunn–Sidak-corrected P < 4 × 10–4 and 3SEQ14), indicating involvement in homologous recombination either directly with identifiable parentals or in their deeper shared evolutionary history—that is, due to shared ancestral recombination events. This is evidence for numerous recombination events occurring in the evolutionary history of the sarbecoviruses22,33; specifying all past events in their correct temporal order34 is challenging and not shown here. Figure 1 (top) shows the distribution of all identified breakpoints (using 3SEQ’s exhaustive triplet search) by the number of candidate recombinant sequences supporting them. The histogram allows for the identification of non-recombining regions (NRRs) by revealing regions with no breakpoints. Sorting these breakpoint-free regions (BFRs) by length results in two segments >5 kb: an ORF1a subregion spanning nucleotides (nt) 3,625–9,150 and the first half of ORF1b spanning nt 13,291–19,628 (sequence numbering given in Source Data, https://github.com/plemey/SARSCoV2origins). Eight other BFRs <500 nt were identified, and the regions were named BFR A–J in order of length. Of the nine breakpoints defining these ten BFRs, four showed phylogenetic incongruence (PI) signals with bootstrap support >80%, adopting previously published criteria on using a combination of mosaic and PI signals to show evidence of past recombination events19. All four of these breakpoints were also identified with the tree-based recombination detection method GARD35.

a, Breakpoints identified by 3SEQ illustrated by percentage of sequences (out of 68) that support a particular breakpoint position. Note that breakpoints can be shared between sequences if they are descendants of the same recombination events. Pink, green and orange bars show BFRs, with region A (nt 13,291–19,628) showing two trimmed segments yielding region A′ (nt 13,291–14,932, 15,405–17,162, 18,009–19,628). Regions B and C span nt 3,625–9,150 and 9,261–11,795, respectively. Concatenated region A′BC is NRR1. Open reading frames are shown above the breakpoint plot, with the variable-loop region indicated in the S protein. b, Similarity plot between SARS-CoV-2 and several selected sequences including RaTG13 (black), SARS-CoV (pink) and two pangolin sequences (orange). The shaded region corresponds to the S protein. c, Maximum likelihood phylogenetic trees rooted on a 2007 virus sampled in Kenya (BtKy72; root truncated from images), shown for five BFRs of the sarbecovirus alignment. Nucleotide positions for phylogenetic inference are 147–695, 962–1,686 (first tree), 3,625–9,150 (second tree, also BFR B), 9,261–11,795 (third tree, also BFR C), 12,443–19,638 (fourth tree) and 23,631–24,633, 24,795–25,847, 27,702–28,843 and 29,574–30,650 (fifth tree). Relevant bootstrap values are shown on branches, and grey-shaded regions show sequences exhibiting phylogenetic incongruence along the genome. S. China corresponds to Guangxi, Yunnan, Guizhou and Guangdong provinces. N. China corresponds to Jilin, Shanxi, Hebei and Henan provinces, and the N. China clade also includes one sequence sampled in Hubei Province in 2004.
The extent of sarbecovirus recombination history can be illustrated by five phylogenetic trees inferred from BFRs or concatenated adjacent BFRs (Fig. 1c). BFRs were concatenated if no phylogenetic incongruence signal could be identified between them. When viewing the last 7 kb of the genome, a clade of viruses from northern China appears to cluster with sequences from southern Chinese provinces but, when inspecting trees from different parts of ORF1ab, the N. China clade is phylogenetically separated from the S. China clade. Individual sequences such as RpShaanxi2011, Guangxi GX2013 and two sequences from Zhejiang Province (CoVZXC21/CoVZC45), as previously shown22,25, have strong phylogenetic recombination signals because they fall on different evolutionary lineages (with bootstrap support >80%) depending on what region of the genome is being examined.
Despite the high frequency of recombination among bat viruses, the block-like nature of the recombination patterns across the genome permits retrieval of a clean subalignment for phylogenetic analysis. Conservatively, we combined the three BFRs >2 kb identified above into non-recombining region 1 (NRR1). Removal of five sequences that appear to be recombinants and two small subregions of BFR A was necessary to ensure that there were no phylogenetic incongruence signals among or within the three BFRs. Alternatively, combining 3SEQ-inferred breakpoints, GARD-inferred breakpoints and the necessity of PI signals for inferring recombination, we can use the 9.9-kb region spanning nucleotides 11,885–21,753 (NRR2) as a putative non-recombining region; this approach is breakpoint-conservative because it is conservative in identifying breakpoints but not conservative in identifying non-recombining regions. Using a third consensus-based approach for identifying recombinant regions in individual sequences—with six different recombination detection methods in RDP5 (ref. 36)—gives a putative recombination-free alignment that we call non-recombinant alignment 3 (NRA3) (see Methods).
All three approaches to removal of recombinant genomic segments point to a single ancestral lineage for SARS-CoV-2 and RaTG13. Two other bat viruses (CoVZXC21 and CoVZC45) from Zhejiang Province fall on this lineage as recombinants of the RaTG13/SARS-CoV-2 lineage and the clade of Hong Kong bat viruses sampled between 2005 and 2007 (Fig. 1c). Specifically, progenitors of the RaTG13/SARS-CoV-2 lineage appear to have recombined with the Hong Kong clade (with inferred breakpoints at 11.9 and 20.8 kb) to form the CoVZXC21/CoVZC45-lineage. Sibling lineages to RaTG13/SARS-CoV-2 include a pangolin sequence sampled in Guangdong Province in March 2019 and a clade of pangolin sequences from Guangxi Province sampled in 2017.
Because the SARS-CoV-2 S protein has been implicated in past recombination events or possibly convergent evolution12, we specifically investigated several subregions of the S protein—the N-terminal domain of S1, the C-terminal domain of S1, the variable-loop region of the C-terminal domain, and S2. The variable-loop region in SARS-CoV-2 shows closer identity to the 2019 pangolin coronavirus sequence than to the RaTG13 bat virus, supported by phylogenetic inference (Fig. 2). On first examination this would suggest that that SARS-CoV-2 is a recombinant of an ancestor of Pangolin-2019 and RaTG13, as proposed by others11,22. However, on closer inspection, the relative divergences in the phylogenetic tree (Fig. 2, bottom) show that SARS-CoV-2 is unlikely to have acquired the variable loop from an ancestor of Pangolin-2019 because these two sequences are approximately 10–15% divergent throughout the entire S protein (excluding the N-terminal domain). It is RaTG13 that is more divergent in the variable-loop region (Extended Data Fig. 1) and thus likely to be the product of recombination, acquiring a divergent variable loop from a hitherto unsampled bat sarbecovirus28. This is notable because the variable-loop region contains the six key contact residues in the RBD that give SARS-CoV-2 its ACE2-binding specificity27,37. These residues are also in the Pangolin Guangdong 2019 sequence. The most parsimonious explanation for these shared ACE2-specific residues is that they were present in the common ancestors of SARS-CoV-2, RaTG13 and Pangolin Guangdong 2019, and were lost through recombination in the lineage leading to RaTG13. This provides compelling support for the SARS-CoV-2 lineage being the consequence of a direct or nearly-direct zoonotic jump from bats, because the key ACE2-binding residues were present in viruses circulating in bats.

SARS-CoV-2 and RaTG13 are the most closely related (their most recent common ancestor nodes denoted by green circles), except in the 222-nt variable-loop region of the C-terminal domain (bar graphs at bottom). In the variable-loop region, RaTG13 diverges considerably with the TMRCA, now outside that of SARS-CoV-2 and the Pangolin Guangdong 2019 ancestor, suggesting that RaTG13 has acquired this region from a more divergent and undetected bat lineage. The genetic distances between SARS-CoV-2 and RaTG13 (bottom) demonstrate that their relationship is consistent across all regions except for the variable loop. The genetic distances between SARS-CoV-2 and Pangolin Guangdong 2019 are consistent across all regions except the N-terminal domain, implying that a recombination event between these two sequences in this region is unlikely. Uncertainty measures are shown in Extended Data Fig. 1. NTD, N-terminal domain; CTD, C-terminal domain.
Ancestry in non-recombinant regions
Using the most conservative approach to identification of a non-recombinant genomic region (NRR1), SARS-CoV-2 forms a sister lineage with RaTG13, with genetically related cousin lineages of coronavirus sampled in pangolins in Guangdong and Guangxi provinces (Fig. 3). Given that these pangolin viruses are ancestral to the progenitor of the RaTG13/SARS-CoV-2 lineage, it is more likely that they are also acquiring viruses from bats. While pangolins could be acting as intermediate hosts for bat viruses to get into humans—they develop severe respiratory disease38 and commonly come into contact with people through trafficking—there is no evidence that pangolin infection is a requirement for bat viruses to cross into humans.

Region A has been shortened to A′ (5,017 nt) based on potential recombination signals within the region. Region B is 5,525 nt long. Sequences are colour-coded by province according to the map. Five example sequences with incongruent phylogenetic positions in the two trees are indicated by dashed lines.
Phylogenies of subregions of NRR1 depict an appreciable degree of spatial structuring of the bat sarbecovirus population across different regions (Fig. 3). One geographic clade includes viruses from provinces in southern China (Guangxi, Yunnan, Guizhou and Guangdong), with its major sister clade consisting of viruses from provinces in northern China (Shanxi, Henan, Hebei and Jilin) as well as Hubei Province in central China and Shaanxi Province in northwestern China. Several of the recombinant sequences in these trees show that recombination events do occur across geographically divergent clades. The Sichuan (SC2018) virus appears to be a recombinant of northern/central and southern viruses, while the two Zhejiang viruses (CoVZXC21 and CoVZC45) appear to carry a recombinant region from southern or central China.
TMRCA for NRRs of SARS-CoV-2 lineage
To avoid artefacts due to recombination, we focused on NRR1 and NRR2 and the recombination-masked alignment NRA3 to infer time-measured evolutionary histories. Visual exploration using TempEst39 indicates that there is no evidence for temporal signal in these datasets (Extended Data Fig. 2). This is not surprising for diverse viral populations with relatively deep evolutionary histories. In such cases, even moderate rate variation among long, deep phylogenetic branches will substantially impact expected root-to-tip divergences over a sampling time range that represents only a small fraction of the evolutionary history40. However, formal testing using marginal likelihood estimation41 does provide some evidence of a temporal signal, albeit with limited log Bayes factor support of 3 (NRR1), 10 (NRR2) and 3 (NRA3); see Supplementary Table 1.
In the absence of a strong temporal signal, we sought to identify a suitable prior rate distribution to calibrate the time-measured trees by examining several coronaviruses sampled over time, including HCoV-OC43, MERS-CoV, and SARS-CoV virus genomes. These datasets were subjected to the same recombination masking approach as NRA3 and were characterized by a strong temporal signal (Fig. 4), but also by markedly different evolutionary rates. Specifically, using a formal Bayesian approach42 (see Methods), we estimate a fast evolutionary rate (0.00169 substitutions per site yr–1, 95% highest posterior density (HPD) interval (0.00131,0.00205)) for SARS viruses sampled over a limited timescale (1 year), a slower rate (0.00078 (0.00063,0.00092) substitutions per site yr–1) for MERS-CoV on a timescale of about 4 years and the slowest rate (0.00024 (0.00019,0.00029) substitutions per site yr–1) for HCoV-OC43 over almost five decades. These differences reflect the fact that rate estimates can vary considerably with the timescale of measurement, a frequently observed phenomenon in viruses known as time-dependent evolutionary rates41,43,44. Over relatively shallow timescales, such differences can primarily be explained by varying selective pressure, with mildly deleterious variants being eliminated more strongly by purifying selection over longer timescales44,45,46. Consistent with this, we estimate a concomitantly decreasing non-synonymous-to-synonymous substitution rate ratio over longer evolutionary timescales: 1.41 (1.20,1.68), 0.35 (0.30,0.41) and 0.133 (0.129,0.136) for SARS, MERS-CoV and HCoV-OC43, respectively. In light of these time-dependent evolutionary rate dynamics, a slower rate is appropriate for calibration of the sarbecovirus evolutionary history. We compare both MERS-CoV- and HCoV-OC43-centred prior distributions (Extended Data Fig. 3) to examine the sensitivity of date estimates to this prior specification.

a–c, Root-to-tip (RtT) divergence as a function of sampling time for the three coronavirus evolutionary histories unfolding over different timescales (HCoV-OC43 (n = 37; a) MERS (n = 35; b) and SARS (n = 69; c)). Decimal years are shown on the x axis for the 1.2 years of SARS sampling in c. d, Mean evolutionary rate estimates plotted against sampling time range for the same three datasets (represented by the same colour as the data points in their respective RtT divergence plots), as well as for the comparable NRA3 using the two different priors for the rate in the Bayesian inference (red points).
We infer time-measured evolutionary histories using a Bayesian phylogenetic approach while incorporating rate priors based on mean MERS-CoV and HCoV-OC43 rates and with standard deviations that allow for more uncertainty than the empirical estimates for both viruses (see Methods). Using both prior distributions, this results in six highly similar posterior rate estimates for NRR1, NRR2 and NRA3, centred around 0.00055 substitutions per site yr–1. The fact that these estimates lie between the rates for MERS-CoV and HCoV-OC43 is consistent with the intermediate sampling time range of about 18 years (Fig. 5). The consistency of the posterior rates for the different prior means also implies that the data do contribute to the evolutionary rate estimate, despite the fact that a temporal signal was visually not apparent (Extended Data Fig. 2). Below, we report divergence time estimates based on the HCoV-OC43-centred rate prior for NRR1, NRR2 and NRA3 and summarize corresponding estimates for the MERS-CoV-centred rate priors in Extended Data Fig. 4. Divergence time estimates based on the HCoV-OC43-centred rate prior for the separate BFRs (Supplementary Table 3) show consistency in TMRCA estimates across the genome.

The time-calibrated phylogeny represents a maximum clade credibility tree inferred for NRR1. Grey tips correspond to bat viruses, green to pangolin, blue to SARS-CoV and red to SARS-CoV-2. The sizes of the black internal node circles are proportional to the posterior node support. 95% credible interval bars are shown for all internal node ages. The inset represents divergence time estimates based on NRR1, NRR2 and NRA3. The boxplots show divergence time estimates (posterior medians) for SARS-CoV-2 (red) and the 2002–2003 SARS-CoV virus (blue) from their most closely related bat virus. Green boxplots show the TMRCA estimate for the RaTG13/SARS-CoV-2 lineage and its most closely related pangolin lineage (Guangdong 2019). Boxplots show interquartile ranges, white lines are medians and box whiskers show the full range of posterior distribution. Transparent bands of interquartile range width and with the same colours are superimposed to highlight the overlap between estimates. In Extended Data Fig. 4 we compare these divergence time estimates to those obtained using the MERS-CoV-centred rate priors for NRR1, NRR2 and NRA3.
The divergence time estimates for SARS-CoV-2 and SARS-CoV from their respective most closely related bat lineages are reasonably consistent among the three approaches we use to eliminate the effects of recombination in the alignment. Using the most conservative approach (NRR1), the divergence time estimate for SARS-CoV-2 and RaTG13 is 1969 (95% HPD: 1930–2000), while that between SARS-CoV and its most closely related bat sequence is 1962 (95% HPD: 1932–1988); see Fig. 5. These are in general agreement with estimates using NRR2 and NRA3, which result in divergence times of 1982 (1948–2009) and 1948 (1879–1999), respectively, for SARS-CoV-2, and estimates of 1952 (1906–1989) and 1970 (1932–1996), respectively, for the divergence time of SARS-CoV from its closest known bat relative. The SARS-CoV divergence times are somewhat earlier than dates previously estimated15 because previous estimates were obtained using a collection of SARS-CoV genomes from human and civet hosts (as well as a few closely related bat genomes), which implies that evolutionary rates were predominantly informed by the short-term SARS outbreak scale and probably biased upwards. Indeed, the rates reported by these studies are in line with the short-term SARS rates that we estimate (Fig. 4). The estimated divergence times for the pangolin virus most closely related to the SARS-CoV-2/RaTG13 lineage range from 1851 (1730–1958) to 1877 (1746–1986), indicating that these pangolin lineages were acquired from bat viruses divergent to those that gave rise to SARS-CoV-2. Current sampling of pangolins does not implicate them as an intermediate host.
Discussion
Identifying the origins of an emerging pathogen can be critical during the early stages of an outbreak, because it may allow for containment measures to be precisely targeted at a stage when the number of daily new infections is still low. Early detection via genomics was not possible during Southeast Asia’s initial outbreaks of avian influenza H5N1 (1997 and 2003–2004) or the first SARS outbreak (2002–2003). By 2009, however, rapid genomic analysis had become a routine component of outbreak response. The 2009 influenza pandemic and subsequent outbreaks of MERS-CoV (2012), H7N9 avian influenza (2013), Ebola virus (2014) and Zika virus (2015) were met with rapid sequencing and genomic characterization. For the current pandemic, the ‘novel pathogen identification’ component of outbreak response delivered on its promise, with viral identification and rapid genomic analysis providing a genome sequence and confirmation, within weeks, that the December 2019 outbreak first detected in Wuhan, China was caused by a coronavirus3. Unfortunately, a response that would achieve containment was not possible. Given what was known about the origins of SARS, as well as identification of SARS-like viruses circulating in bats that had binding sites adapted to human receptors29,30,31, appropriate measures should have been in place for immediate control of outbreaks of novel coronaviruses. The key to successful surveillance is knowing which viruses to look for and prioritizing those that can readily infect humans47.
The difficulty in inferring reliable evolutionary histories for coronaviruses is that their high recombination rate48,49 violates the assumption of standard phylogenetic approaches because different parts of the genome have different histories. To begin characterizing any ancestral relationships for SARS-CoV-2, NRRs of the genome must be identified so that reliable phylogenetic reconstruction and dating can be performed. Evolutionary rate estimation can be profoundly affected by the presence of recombination50. Because there is no single accepted method of inferring breakpoints and identifying clean subregions with high certainty, we implemented several approaches to identifying three classic statistical signals of recombination: mosaicism, phylogenetic incongruence and excessive homoplasy51. Our most conservative approach attempted to ensure that putative NRRs had no mosaic or phylogenetic incongruence signals. A second breakpoint-conservative approach was conservative with respect to breakpoint identification, but this means that it is accepting of false-negative outcomes in breakpoint inference, resulting in less certainty that a putative NRR truly contains no breakpoints. A third approach attempted to minimize the number of regions removed while also minimizing signals of mosaicism and homoplasy. The origins we present in Fig. 5 (NRR1) are conservative in the sense that NRR1 is more likely to be non-recombinant than NRR2 or NRA3. Because the estimated rates and divergence dates were highly similar in the three datasets analysed, we conclude that our estimates are robust to the method of identifying a genome’s NRRs.
Due to the absence of temporal signal in the sarbecovirus datasets, we used informative prior distributions on the evolutionary rate to estimate divergence dates. Calibration of priors can be performed using other coronaviruses (SARS-CoV, MERS-CoV and HCoV-OC43), but estimated rates vary with the timescale of sample collection. In the presence of time-dependent rate variation, a widely observed phenomenon for viruses43,44,52, slower prior rates appear more appropriate for sarbecoviruses that currently encompass a sampling time range of about 18 years. Our approach resulted in similar posterior rates using two different prior means, implying that the sarbecovirus data do inform the rate estimate even though a root-to-tip temporal signal was not apparent.
The relatively fast evolutionary rate means that it is most appropriate to estimate shallow nodes in the sarbecovirus evolutionary history. Accurate estimation of ages for deeper nodes would require adequate accommodation of time-dependent rate variation. While such models have recently been made available, we lack the information to calibrate the rate decline over time (for example, through internal node calibrations44). As a proxy, it would be possible to model the long-term purifying selection dynamics as a major source of time-dependent rates43,44,52, but this is beyond the scope of the current study. The assumption of long-term purifying selection would imply that coronaviruses are in endemic equilibrium with their natural host species, horseshoe bats, to which they are presumably well adapted. While there is evidence of positive selection in the sarbecovirus lineage leading to RaTG13/SARS-CoV-2 (ref. 53), this is inferred to have occurred before the divergence of RaTG13 and SARS-CoV-2 and thus should not influence our inferences.
Of importance for future spillover events is the appreciation that SARS-CoV-2 has emerged from the same horseshoe bat subgenus that harbours SARS-like coronaviruses. Another similarity between SARS-CoV and SARS-CoV-2 is their divergence time (40–70 years ago) from currently known extant bat virus lineages (Fig. 5). This long divergence period suggests there are unsampled virus lineages circulating in horseshoe bats that have zoonotic potential due to the ancestral position of the human-adapted contact residues in the SARS-CoV-2 RBD. Without better sampling, however, it is impossible to estimate whether or how many of these additional lineages exist. While there is involvement of other mammalian species—specifically pangolins for SARS-CoV-2—as a plausible conduit for transmission to humans, there is no evidence that pangolins are facilitating adaptation to humans. A hypothesis of snakes as intermediate hosts of SARS-CoV-2 was posited during the early epidemic phase54, but we found no evidence of this55,56; see Extended Data Fig. 5.
With horseshoe bats currently the most plausible origin of SARS-CoV-2, it is important to consider that sarbecoviruses circulate in a variety of horseshoe bat species with widely overlapping species ranges57. Nevertheless, the viral population is largely spatially structured according to provinces in the south and southeast on one lineage, and provinces in the centre, east and northeast on another (Fig. 3). This boundary appears to be rarely crossed. Two exceptions can be seen in the relatively close relationship of Hong Kong viruses to those from Zhejiang Province (with two of the latter, CoVZC45 and CoVZXC21, identified as recombinants) and a recombinant virus from Sichuan for which part of the genome (region B of SC2018 in Fig. 3) clusters with viruses from provinces in the centre, east and northeast of China. SARS-CoV-2 and RaTG13 are also exceptions because they were sampled from Hubei and Yunnan, respectively. The fact that they are geographically relatively distant is in agreement with their somewhat distant TMRCA, because the spatial structure suggests that migration between their locations may be uncommon. From this perspective, it may be useful to perform surveillance for more closely related viruses to SARS-CoV-2 along the gradient from Yunnan to Hubei.
It is clear from our analysis that viruses closely related to SARS-CoV-2 have been circulating in horseshoe bats for many decades. The unsampled diversity descended from the SARS-CoV-2/RaTG13 common ancestor forms a clade of bat sarbecoviruses with generalist properties—with respect to their ability to infect a range of mammalian cells—that facilitated its jump to humans and may do so again. Although the human ACE2-compatible RBD was very likely to have been present in a bat sarbecovirus lineage that ultimately led to SARS-CoV-2, this RBD sequence has hitherto been found in only a few pangolin viruses. Furthermore, the other key feature thought to be instrumental in the ability of SARS-CoV-2 to infect humans—a polybasic cleavage site insertion in the S protein—has not yet been seen in another close bat relative of the SARS-CoV-2 virus.
The existing diversity and dynamic process of recombination amongst lineages in the bat reservoir demonstrate how difficult it will be to identify viruses with potential to cause major human outbreaks before they emerge. This underscores the need for a global network of real-time human disease surveillance systems, such as that which identified the unusual cluster of pneumonia in Wuhan in December 2019, with the capacity to rapidly deploy genomic tools and functional studies for pathogen identification and characterization.
MethodsDataset compilationSARBECOVIRUS DATA
Complete genome sequence data were downloaded from GenBank and ViPR; accession numbers of all 68 sequences are available in Supplementary Table 4. Sequences were aligned by MAFTT58 v.7.310, with a final alignment length of 30,927, and used in the analyses below.
HCOV-OC43
We compiled a dataset including 27 human coronavirus OC43 virus genomes and ten related animal virus genomes (six bovine, three white-tailed deer and one canine virus). The canine viral genome was excluded from the Bayesian phylogenetic analyses because temporal signal analyses (see below) indicated that it was an outlier.
MERS-COV
We extracted a similar number (n = 35) of genomes from a MERS-CoV dataset analysed by Dudas et al.59 using the phylogenetic diversity analyser tool60 (v.0.5).
SARS-COV
We compiled a set of 69 SARS-CoV genomes including 58 sampled from humans and 11 sampled from civets and raccoon dogs. This dataset comprises an updated version of that used in Hon et al.15 and includes a cluster of genomes sampled in late 2003 and early 2004, but the evolutionary rate estimate without this cluster (0.00175 substitutions per site yr–1 (0.00117,0.00229)) is consistent with the complete dataset (0.00169 substitutions per site yr–1, (0.00131,0.00205)).
Sarbecovirus, HCoV-OC43 and SARS-CoV data were assembled from GenBank to be as complete as possible, with sampling year as an inclusion criterion. MERS-CoV data were subsampled to match sample sizes with SARS-CoV and HCoV-OC43.
Recombination analysis
Because coronaviruses are known to be highly recombinant, we used three different approaches to identify non-recombinant regions for use in our Bayesian time-calibrated phylogenetic inference.
First, we took an approach that relies on identification of mosaic regions (via 3SEQ14 v.1.7) that are also supported by PI signals19. Because 3SEQ is the most statistically powerful of the mosaic methods61, we used it to identify the best-supported breakpoint history for each potential child (recombinant) sequence in the dataset. A single 3SEQ run on the genome alignment resulted in 67 out of 68 sequences supporting some recombination in the past, with multiple candidate breakpoint ranges listed for each putative recombinant. Next, we (1) collected all breakpoints into a single set, (2) complemented this set to generate a set of non-breakpoints, (3) grouped non-breakpoints into contiguous BFRs and (4) sorted these regions by length. A phylogenetic tree—using RAxML v8.2.8 (ref. 62,63), the GTR + Γ model and 100 bootstrap replicates—was inferred for each BFR >500 nt.
We considered (1) the possibility that BFRs could be combined into larger non-recombinant regions and (2) the possibility of further recombination within each BFR.
We named the length-sorted BFRs as: BFR A (nt positions 13,291–19,628, length = 6,338 nt), BFR B (nt positions 3,625–9,150, length = 5,526 nt), BFR C (nt positions 9,261–11,795, length = 2,535 nt), BFR D (nt positions 27,702–28,843, length = 1,142 nt) and six further regions (E–J). Phylogenetic trees and exact breakpoints for all ten BFRs are shown in Supplementary Figs. 1–10. Regions A–C had similar phylogenetic relationships among the southern China bat viruses (Yunnan, Guangxi and Guizhou provinces), the Hong Kong viruses, northern Chinese viruses (Jilin, Shanxi, Hebei and Henan provinces, including Shaanxi), pangolin viruses and the SARS-CoV-2 lineage. Because these subclades had different phylogenetic relationships in region D (Supplementary Fig. 4), that region and shorter BFRs were not included in combined putative non-recombinant regions.
Regions A–C were further examined for mosaic signals by 3SEQ, and all showed signs of mosaicism. In region A, we removed subregion A1 (nt positions 3,872–4,716 within region A) and subregion A4 (nt 1,642–2,113) because both showed PI signals with other subregions of region A. After removal of A1 and A4, we named the new region A′. In addition, sequences NC_014470 (Bulgaria 2008), CoVZXC21, CoVZC45 and DQ412042 (Hubei-Yichang) needed to be removed to maintain a clean non-recombinant signal in A′. Region B showed no PI signals within the region, except one including sequence SC2018 (Sichuan), and thus this sequence was also removed from the set. Region C showed no PI signals within it. Combining regions A′, B and C and removing the five named sequences gives us putative NRR1, as an alignment of 63 sequences. We say that this approach is conservative because sequences and subregions generating recombination signals have been removed, and BFRs were concatenated only when no PI signals could be detected between them. The construction of NRR1 is the most conservative as it is least likely to contain any remaining recombination signals.
In our second stage, we wanted to construct non-recombinant regions where our approach to breakpoint identification was as conservative as possible. We call this approach breakpoint-conservative, but note that this has the opposite effect to the construction of NRR1 in that this approach is the most likely to allow breakpoints to remain inside putative non-recombining regions. In other words, a true breakpoint is less likely to be called as such (this is breakpoint-conservative), and thus the construction of a non-recombining region may contain true recombination breakpoints (with insufficient evidence to call them as such). In this approach, we considered a breakpoint as supported only if it had three types of statistical support: from (1) mosaic signals identified by 3SEQ, (2) PI signals identified by building trees around 3SEQ’s breakpoints and (3) the GARD algorithm35, which identifies breakpoints by identifying PI signals across proposed breakpoints. Because 3SEQ identified ten BFRs >500 nt, we used GARD’s (v.2.5.0) inference on 10, 11 and 12 breakpoints. A reduced sequence set of 25 sequences chosen to capture the breadth of diversity in the sarbecoviruses (obvious recombinants not involving the SARS-CoV-2 lineage were also excluded) was used because GARD is computationally intensive. GARD identified eight breakpoints that were also within 50 nt of those identified by 3SEQ. PI signals were identified (with bootstrap support >80%) for seven of these eight breakpoints: positions 1,684, 3,046, 9,237, 11,885, 21,753, 22,773 and 24,628. Using these breakpoints, the longest putative non-recombining segment (nt 1,885–21,753) is 9.9 kb long, and we call this region NRR2.
Our third approach involved identifying breakpoints and masking minor recombinant regions (with gaps, which are treated as unobserved characters in probabilistic phylogenetic approaches). Specifically, we used a combination of six methods implemented in v.5.5 of RDP5 (ref. 36) (RDP, GENECONV, MaxChi, Bootscan, SisScan and 3SEQ) and considered recombination signals detected by more than two methods for breakpoint identification. Except for specifying that sequences are linear, all settings were kept to their defaults. Based on the identified breakpoints in each genome, only the major non-recombinant region is kept in each genome while other regions are masked. To evaluate the performance procedure, we confirmed that the recombination masking resulted in (1) a markedly different outcome of the PHI test64, (2) removal of well-supported (bootstrap value >95%) incompatible splits in Neighbor-Net65 and (3) a near-complete reduction of mosaic signal as identified by 3SEQ. If the latter still identified non-negligible recombination signal, we removed additional genomes that were identified as major contributors to the remaining signal. This produced non-recombining alignment NRA3, which included 63 of the 68 genomes.
Bayesian divergence time estimation
We focused on these three non-recombining regions/alignments for divergence time estimation; this avoids inappropriate modelling of evolutionary processes with recombination on strictly bifurcating trees, which can result in different artefacts such as homoplasies that inflate branch lengths and lead to apparently longer evolutionary divergence times. To examine temporal signal in the sequenced data, we plotted root-to-tip divergence against sampling time using TempEst39 v.1.5.3 based on a maximum likelihood tree. The latter was reconstructed using IQTREE66 v.2.0 under a general time-reversible (GTR) model with a discrete gamma distribution to model inter-site rate variation.
Time-measured phylogenetic reconstruction was performed using a Bayesian approach implemented in BEAST42 v.1.10.4. When the genomic data included both coding and non-coding regions we used a single GTR + Γ substitution model; for concatenated coding genes we partitioned the alignment by codon position and specified an independent GTR + Γ model for each partition with a separate gamma model to accommodate inter-site rate variation. We used an uncorrelated relaxed clock model with log-normal distribution for all datasets, except for the low-diversity SARS data for which we specified a strict molecular clock model. For the HCoV-OC43, MERS-CoV and SARS datasets we specified flexible skygrid coalescent tree priors. In the absence of any reasonable prior knowledge on the TMRCA of the sarbecovirus datasets (which is required for grid specification in a skygrid model), we specified a simpler constant size population prior. As informative rate priors for the analysis of the sarbecovirus datasets, we used two different normal prior distributions: one with a mean of 0.00078 and s.d. = 0.00075 and one with a mean of 0.00024 and s.d. = 0.00025. These means are based on the mean rates estimated for MERS-CoV and HCoV-OC43, respectively, while the standard deviations are set ten times higher than empirical values to allow greater prior uncertainty and avoid strong bias (Extended Data Fig. 3). In our analyses of the sarbecovirus datasets, we incorporated the uncertainty of the sampling dates when exact dates were not available. To estimate non-synonymous over synonymous rate ratios for the concatenated coding genes, we used the empirical Bayes Renaissance counting’procedure67. Temporal signal was tested using a recently developed marginal likelihood estimation procedure41 (Supplementary Table 1).
Posterior distributions were approximated through Markov chain Monte Carlo sampling, which were run sufficiently long to ensure effective sampling sizes >100. BEAST inferences made use of the BEAGLE v.3 library68 for efficient likelihood computations. We used TreeAnnotator to summarize posterior tree distributions and annotated the estimated values to a maximum clade credibility tree, which was visualized using FigTree.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequence data analysed in this manuscript are available at https://github.com/plemey/SARSCoV2origins. Note that six of these sequences fall under the terms of use of the GISAID platform.
| ||
| Analysis: Has COVID-19 been around for 70 years? | ||
FILE - This undated electron microscope image made available by the U.S. National Institutes of Health in February 2020 shows the Novel Coronavirus SARS-CoV-2, yellow, emerging from the surface of cells, pink, cultured in the lab. Also known as 2019-nCoV, the virus causes COVID-19. The sample was isolated from a patient in the U.S. On Friday, May 29, 2020, The Associated Press reported on stories circulating online incorrectly asserting that coronavirus has an HIV protein that proves it was genetically modified. Experts say the coronavirus has no HIV sequences in it’s genetic makeup. Since the early days of the coronavirus outbreak, social media posts have tried to cast doubt on its origins. (NIAID-RML via AP)
less
FILE - This undated electron microscope image made available by the U.S. National Institutes of Health in February 2020 shows the Novel Coronavirus SARS-CoV-2, yellow, emerging from the surface of cells, pink,
... more
Analysis: Has COVID-19 been around for 70 years?
Has the coronavirus been around for decades? Maybe, according to a consortium of researchers from the United States, China and Europe.
It has proven difficult to pick apart the genetic lineage of the virus. But, now an international team of scientists say they’ve done it, and found clues (and warnings) about both the history of this virus and the presence of its relatives.
The problem, as was explained in a release from the University of Glasgow, is that the coronavirus is “highly recombinant, meaning different regions of the virus’s genome can be derived from multiple sources,” said Maciej Boni, associate professor of biology at Penn State. “This has made it difficult to reconstruct SARS-CoV-2’s origins. You have to identify all the regions that have been recombining and trace their histories.”
The research findings, which were published yesterday in the journal Nature Microbiology, suggest that the virus that causes COVID-19 may have been circulating among bat populations for as much as 70 years.
Importantly, one aspect of the virus (the part that allows the virus to identify and bind to human cells) is common to other, similar viruses. That’s … not a good thing, according to David L. Robertson, professor of computational virology, MRC-University of Glasgow Centre for Virus Research.
“This means that other viruses that are capable of infecting humans are circulating in horseshoe bats in China,” he said.
The good news is that another feature of the virus that allows it to infect human hosts “has not yet been seen in another close bat relative of the SARS-CoV-2 virus,” according to Robertson.
The bad news is, this research suggests that more pandemics are likely.
“We were too late in responding to the initial SARS-CoV-2 outbreak, but this will not be our last coronavirus pandemic,” Boni said.
| ||
| Analysis: Has COVID-19 been around for 70 years? | ||
But, now an international team of scientists say they've done it, and found clues (and warnings) about both the history of this virus and the presence of ... | ||
| Google Alert - Coronavirus in health care workers: COVID-19 staff reductions | ||
Dentists, physicians and other health care practitioners have had their livelihoods hit hardest in the health care sector. About 243,000 jobs were lost in ... | ||
| Researchers identify evolutionary origins of SARS-CoV-2 | ||
By reconstructing the evolutionary history of SARS-CoV-2, the virus that is responsible for the COVID-19 pandemic, an international research team of Chinese, European and U.S. scientists has discovered that the lineage that gave rise to the virus has been circulating in bats for decades and likely includes other viruses with the ability to infect humans. The findings have implications for the prevention of future pandemics stemming from this lineage.
“Coronaviruses have genetic material that is highly recombinant, meaning different regions of the virus’s genome can be derived from multiple sources,” said Maciej Boni, associate professor of biology, Penn State. “This has made it difficult to reconstruct SARS-CoV-2’s origins. You have to identify all the regions that have been recombining and trace their histories. To do that, we put together a diverse team with expertise in recombination, phylogenetic dating, virus sampling, and molecular and viral evolution.”
The team used three different bioinformatic approaches to identify and remove the recombinant regions within the SARS-CoV-2 genome. Next, they reconstructed phylogenetic histories for the non-recombinant regions and compared them to each other to see which specific viruses have been involved in recombination events in the past. They were able to reconstruct the evolutionary relationships between SARS-CoV-2 and its closest known bat and pangolin viruses. Their findings appear today (July 28) in Nature Microbiology.
The researchers found that the lineage of viruses to which SARS-CoV-2 belongs diverged from other bat viruses about 40-70 years ago. Importantly, although SARS-CoV-2 is genetically similar (about 96%) to the RaTG13 coronavirus, which was sampled from a Rhinolophus affinis horseshoe bat in 2013 in Yunnan province, China, the team found that it diverged from RaTG13 a relatively long time ago, in 1969.
“The ability to estimate divergence times after disentangling recombination histories, which is something we developed in this collaboration, may lead to insights into the origins of many different viral pathogens,” said Philippe Lemey, principal investigator in the Department of Evolutionary and Computational Virology, KE Leuven.
The team found that one of the older traits that SARS-CoV-2 shares with its relatives is the receptor-binding domain (RBD) located on the Spike protein, which enables the virus to recognize and bind to receptors on the surfaces of human cells.
“This means that other viruses that are capable of infecting humans are circulating in horseshoe bats in China,” said David L. Robertson, professor of computational virology, MRC-University of Glasgow Centre for Virus Research.
Will these viruses be capable of jumping directly from bats into humans or will an intermediate species be required to make the leap? According to Robertson, for SARS-CoV-2, other research groups incorrectly proposed that key evolutionary changes occurred in pangolins.
“SARS-CoV-2’s RBD sequence has so far only been found in a few pangolin viruses,” said Robertson. “Furthermore, the other key feature thought to be instrumental to SARS-CoV-2’s ability to infect humans — a polybasic cleavage site insertion in the Spike protein — has not yet been seen in another close bat relative of the SARS-CoV-2 virus. Yet, while it is possible that pangolins may have acted as an intermediate host facilitating transmission of SARS-CoV-2 to humans, no evidence exists to suggest that pangolin infection is a requirement for bat viruses to cross into humans. Instead, our research suggests that SARS-CoV-2 likely evolved the ability to replicate in the upper respiratory tract of both humans and pangolins.”
The team concluded that preventing future pandemics will require better sampling within wild bats and the implementation of human disease surveillance systems that are able to identify novel pathogens in humans and respond in real time.
“The key to successful surveillance,” said Robertson, “is knowing which viruses to look for and prioritizing those that can readily infect humans. We should have been better prepared for a second SARS virus.”
Boni added, “We were too late in responding to the initial SARS-CoV-2 outbreak, but this will not be our last coronavirus pandemic. A much more comprehensive and real-time surveillance system needs to be put in place to catch viruses like this when case numbers are still in the double digits.”
Other authors on the paper include: Xiaowei Jiang, lecturer in bioinformatics, Xi’an Jiaotong-Liverpool University; Tommy Tsan-Yuk Lam, assistant professor of public health, University of Hong Kong; Blair Perry, graduate student, University of Texas Arlington; Todd Castoe, associate professor of biology, University of Texas Arlington; and Andrew Rambaut, professor of molecular evolution, Institute of Evolutionary Biology, University of Edinburgh.
Support for this research was provided by the European Research Council, the Medical Research Council, the Research Foundation — Flanders and the National Natural Science Foundation of China.
| ||
| Google Alert - new york times on coronavirus: Mysterious Coronavirus Outbreak Catches Vietnam by Surprise | ||
The surge of the coronavirus in Vietnam, which has so far recorded fewer than 450 cases ... In the Australian state of Victoria, authorities announced 295 new cases on ... A waiter at a pizzeria in Hanoi tested positive for the coronavirus after ... Photo Illustration by The New York Times; Google Street View ... | ||
| Google Alert - Coronavirus and cyber attacks: Report Shows COVID-19 Five Times More Disruptive Than Cyberattacks | ||
In 2020, the pandemic has emerged as the most potent threat to economic growth, more than trade sanctions, natural disasters and cyberattacks ... | ||
| mikenov on Twitter: The Disease-X-19: 10:44 AM 7/29/2020 Is this the sign of the rati diseasex19.blogspot.com/2020/07/1044-a | ||
The Disease-X-19: 10:44 AM 7/29/2020 – Is this the sign of the “rati… diseasex19.blogspot.com/2020/07/1044-a…
| ||
| Google Alert - Sars-Cov-2 origins: Gene Study Shows How Coronavirus Swept Through the Diamond Princess | ||
In the new study, the Japanese team sought to determine the origins of the cruise ship outbreak. To do so, they analyzed the DNA of samples of SARS- ... | ||
| Covid-19 as multi-organ and multi-system disease - Google Search google.com/search?q=Covid | ||
| The Disease-X-19: 10:44 AM 7/29/2020 - Is this the sign of the "rati... diseasex19.blogspot.com/2020/07/1044-a | ||
The Disease-X-19: 10:44 AM 7/29/2020 - Is this the sign of the "rati... diseasex19.blogspot.com/2020/07/1044-a
| ||
| Polls shows dead even race in Georgia between Trump, Biden; GOP leads Senate races | ||
Its been nearly three decades since a Democrat won Georgia in a presidential election. But a new poll in Georgia indicates the states 16 electoral votes are up for grabs in the race between President Trump and Democratic challenger Joe Biden.   | ||
| US Officials: Russia Behind Spread of Virus Disinformation - gvwire.com | ||
US Officials: Russia Behind Spread of Virus Disinformation gvwire.com | ||
| FOX News: Trump says Harris would be fine choice as Biden running mate | ||
President Trump is weighing in on one of the likely top contenders Democratic challenger Joe Bidens considering as his running mate  | ||
| Deutsche Welle from Michael_Novakhov (6 sites): Deutsche Welle: Tortured and abused the harrowing experiences of North Korean women prisoners | ||
Pyongyang has dismissed the UN report, which quotes 100 women who experienced torture, malnutrition, forced labor, sexual violence and murder of new-born children. Julian Ryall reports. | ||
| FOX News: US announces plans to remove 12,000 troops from Germany, with half coming home | ||
The U.S. has announced plans Wednesday to shift 12,000 troops out of Germany as part of a multibillion-dollar effort to deter Russian influence and reassure European allies in the region.   | ||
| 10:44 AM 7/29/2020 - Is this the sign of the "rational" (lab, man-made) rather than the "natural" design of this infection(s) and its pathogen(s)? | ||
 10:44 AM 7/29/2020 Is this the sign of the "rational" (lab, man-made) rather than the "natural" design of this infection(s) and its pathogen(s)? "the increased infectivity of the G-form is likely due to a higher rate of profitable binding encounters with the host receptor." - The SARS-CoV-2 Spike Variant D614G Favors an Open Conformational State
Saved and Shared Stories from Michael_Novakhov | Page Headlines | Saved and Shared Stories In 250 Brief Posts ___________________________________________________________________ Michael Novakhov - SharedNewsLinks - Latest Post | ||
| Emergent SARS-CoV-2 D614G spike mutation more infective due to "open" morphology | ||
Researchers from the Los Alamos National Laboratory and Duke Human Vaccine Institute & Department of Surgery demonstrated that globally prevalent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with the amino acid substitution D614G in the spike protein is much more infective due to profitable binding encounters with the host receptors. Their findings are currently available on the bioRxiv* preprint server.
Coronavirus disease (COVID-19) caused by SARS-CoV-2 represents a salient global health emergency. This virus infects human cells by utilizing the receptor-binding domain (RBD) of the trimeric spike protein to bind to the angiotensin-converting enzyme 2 (ACE2) receptor found in many cells of the human body.
Amidst the COVID-19 pandemic, the emergence of a SARS-CoV-2 variant, which carried the amino acid substitution D614G in the spike protein, has been observed. Very soon, that exact viral strain became globally prevalent.
Mechanistically speaking, in the "all-down" spike conformation, all three RBDs of the protomers are in the closed orientation. However, binding of the RBD to ACE2 necessitates the transition of one of the protomers from the closed to an open conformation.
Such an infection-capable configuration is referred to as open or "one-up" spike conformation. Consequently, the resulting G-form is considered more infectious in the laboratory conditions and associated with increased viral loads in infected people.
To identify the factors that lead to such distinct viral dynamics, a research group from the Los Alamos National Laboratory and Duke Human Vaccine Institute & Department of Surgery in the United States conducted a meticulous mechanistic analysis and obtained some interesting and novel insights.
 The Spike complex is shown in the “all-down” conformation. Its S1 and S2 subunits are depicted in red and blue. (B) The Spike complex is shown in the “one-up” conformation. It is composed of three protomers, with the L-down protomer depicted in purple, the Up protomer in orange, and the R-down protomer in cyan. The dashed white circle indicates a D614G site. Here, only one of the three D614G sites are shown. (C) Definition of twelve domains and their residues used for analysis of simulations. It was necessary to define domains for every region for analysis, so some regions may have arbitrarily assigned names or not follow canonical sequence ranges. We display the domains highlighted in the Spike structure, shown from two different perspectives. Definitions of domain abbreviations: NTD, N-Terminal Domain; CT0, C313 Terminal domain 0; RBD, Receptor Binding Domain; CT1, C-Terminal domain 1; CT2, C-Terminal domain 2; S2S2’, S1/S2 Cleavage to S2’; FP, Fusion Peptide; FPR, Fusion Peptide Region; HR1, Heptad Repeat 1; CH, Center Helix; CD, Connector Domain; and CD1, Connector Domain 1. Images in (A)-(B) were created with PyMol (15); images in (C) were prepared using VMD (16).")
Structural Representation of the Spike protein. (A) The Spike complex is shown in the “all-down” conformation. Its S1 and S2 subunits are depicted in red and blue. (B) The Spike complex is shown in the “one-up” conformation. It is composed of three protomers, with the L-down protomer depicted in purple, the Up protomer in orange, and the R-down protomer in cyan. The dashed white circle indicates a D614G site. Here, only one of the three D614G sites are shown. (C) Definition of twelve domains and their residues used for analysis of simulations. It was necessary to define domains for every region for analysis, so some regions may have arbitrarily assigned names or not follow canonical sequence ranges. We display the domains highlighted in the Spike structure, shown from two different perspectives. Definitions of domain abbreviations: NTD, N-Terminal Domain; CT0, C313 Terminal domain 0; RBD, Receptor Binding Domain; CT1, C-Terminal domain 1; CT2, C-Terminal domain 2; S2S2’, S1/S2 Cleavage to S2’; FP, Fusion Peptide; FPR, Fusion Peptide Region; HR1, Heptad Repeat 1; CH, Center Helix; CD, Connector Domain; and CD1, Connector Domain 1. Images in (A)-(B) were created with PyMol (15); images in (C) were prepared using VMD (16).
Appraising molecular-level impacts
In a nutshell, this study successfully used multifarious replicas of microsecond all-atom simulations in order to probe the molecular-level impact of the amino acid substitution D614G on closed and open states of the SARS-CoV-2 spike protein.
Hydrogen bonding interactions of the speculated critical residues were studied in-depth, primarily to observe whether hydrogen bond reduction can lead to a conformational relaxation of the spike protein.
Moreover, a global differential contact analysis was also pursued, and the researchers have modeled the impact of highly-flexible glycan ensembles on RBD exposure and epitope shielding with the use of a quantitative measure known as the glycan encounter factor.
A higher rate of profitable binding encounters
More specifically, the G614 form was shown to lead to a rearrangement in residue-residue contacts and hydrogen bonding that would lead to an increased population of the one-up spike ensemble or open conformations.
Likewise, a careful analysis of RBD exposure (including the role of highly-flexible glycans) revealed that both the ACE2 binding site and critical neutralizing antibody epitopes in the RBD are definitely more accessible in the one upstate.
As a consequence, the increased infectivity of the aforementioned G-form can be observed, likely arising due to a much higher rate of profitable binding encounters with the receptor located at the host cells.
 Surface representation of the all-down Spike complex. Colors ranging between red, white and blue represent values for the glycan encounter factor (GEF). GEF describes relative exposure and coverage of protein surfaces. (B) Surface representation of the one-up Spike. Colors ranging between cyan, white, and magenta indicate GEF differences between the closed and open structures. The RBD in the up orientation is indicated by the dotted circle, where the originally buried (magenta) region around 458-478 is now exposed. (C-D) Top view of the Spike protein apex in (C) all-down and (D) one-up conformations. Protein surface is shown in grey. Glycan ensembles covering the protein surface are represented by point densities. Fucosylated 2 and 3 antennae complex (FA2 and FA3) glycans and oligomannose (OM) glycans are depicted in dark maroon, orange, and green. Binding site region, which includes receptor binding motif (residues 438-506) and C105 antibody binding residues (i.e. 403, 405, 406, 408, 409, 415-417, 420, 421, 449, 453, 455-460, 473-477, 486, 487, 489, 493-496, 498, 500-505), is colored yellow. These binding site residues are significantly more exposed in the one-up conformation, compared to the all-down case. (E) Residue-wise GEF of the RBD domain. Buried residues are colored blue. Exposure increases as the color changes toward white and red.")
RBD is significantly more exposed in the one-up conformation. (A) Surface representation of the all-down Spike complex. Colors ranging between red, white and blue represent values for the glycan encounter factor (GEF). GEF describes relative exposure and coverage of protein surfaces. (B) Surface representation of the one-up Spike. Colors ranging between cyan, white, and magenta indicate GEF differences between the closed and open structures. The RBD in the up orientation is indicated by the dotted circle, where the originally buried (magenta) region around 458-478 is now exposed. (C-D) Top view of the Spike protein apex in (C) all-down and (D) one-up conformations. Protein surface is shown in grey. Glycan ensembles covering the protein surface are represented by point densities. Fucosylated 2 and 3 antennae complex (FA2 and FA3) glycans and oligomannose (OM) glycans are depicted in dark maroon, orange, and green. Binding site region, which includes receptor binding motif (residues 438-506) and C105 antibody binding residues (i.e. 403, 405, 406, 408, 409, 415-417, 420, 421, 449, 453, 455-460, 473-477, 486, 487, 489, 493-496, 498, 500-505), is colored yellow. These binding site residues are significantly more exposed in the one-up conformation, compared to the all-down case. (E) Residue-wise GEF of the RBD domain. Buried residues are colored blue. Exposure increases as the color changes toward white and red.
Change towards a higher one-up state population
This means that both ACE2 binding and RBD-targeting antibody binding are expected to increase in the one-up state; hence, a change towards a higher one-up state population probably represents a dominant effect of the D614G substitution.
Accordingly, such form is also predicted to be much more sensitive to neutralization due to enhanced exposure of the RBD, which is a primary target region for neutralizing antibodies.
In any case, the structural data and mechanistic studies presented here, the enhanced neutralizing antibody sensitivity, as well as the experimentally determined increase in infectivity make a rather consistent story with possible therapeutic implications.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
| ||
| The SARS-CoV-2 Spike Variant D614G Favors an Open Conformational State | bioRxiv | ||
Summary
The COVID-19 pandemic underwent a rapid transition with the emergence of a SARS-CoV-2 variant that carried the amino acid substitution D614G in the Spike protein that became globally prevalent. The G-form is both more infectious in vitro and associated with increased viral loads in infected people. To gain insight into the mechanism underlying these distinctive characteristics, we employed multiple replicas of microsecond all-atom simulations to probe the molecular-level impact of this substitution on Spike’s closed and open states. The open state enables Spike interactions with its human cellular receptor, ACE2. Here we show that changes in the inter-protomer energetics due to the D614G substitution favor a higher population of infection-capable (open) states. The inter-protomer interactions between S1 and S2 subunits in the open state of the D-form are asymmetric. This asymmetry is resolved in the G-form due to the release of tensile hydrogen bonds resulting in an increased population of open conformations. Thus, the increased infectivity of the G-form is likely due to a higher rate of profitable binding encounters with the host receptor. It is also predicted to be more neutralization sensitive due to enhanced exposure of the receptor binding domain, a key target region for neutralizing antibodies.
| ||
| Two new studies suggest 'the plot thickening' | ||
Signed in as Michael_Novakhov
Send this story to NewsBlur

Shared stories are on their way...
| ||
| Covid-19 and the heart: two new studies offer insights - CNN | ||
Covid-19's impact on the heart: Two new studies suggest 'the plot thickening'(16 Videos)
(CNN)Since the coronavirus pandemic first began, evidence has emerged showing that Covid-19 can damage more than the lungs.
The disease caused by the novel coronavirus can harm other organs in the body -- including the heart -- and now two separate studies, published in the journal JAMA Cardiology on Monday, provide more insight into how Covid-19 may have a prolonged impact on heart health in those who have recovered from illness and may have caused cardiac infection in those who died.
 MUST WATCH
Doctors baffled as virus triggers mysterious health issues 02:47
"We've understood for a few months now that Covid-19 is not only a respiratory infection but a multi-system infection," said cardiologist Dr. Nieca Goldberg, medical director of the NYU Women's Heart Program and senior adviser for women's health strategy at NYU Langone Health in New York, who was not involved in either study.
"There is an acute inflammatory response, increased blood clotting and cardiac involvement. And the cardiac involvement can either be due to direct involvement of the heart muscle by the infection and its inflammatory response. It could be due to blood clots that are formed, causing an obstruction of arteries," Goldberg said.
Content by CNN Underscored
The best deals in Apple’s Amazon store
Yes, Apple has set up shop on Amazon. That means you can get official Apple products with free Prime shipping.
"Sometimes people have very fast heart rates that can, over time, weaken the heart muscle, reduce the heart muscle function. So there are multiple ways during this infection that it can involve the heart."
Read More
Inflammation of the heart
One of the JAMA Cardiology studies found that, among 100 adults who recently recovered from Covid, 78% showed some type of cardiac involvement in MRI scans and 60% had ongoing inflammation in the heart.
The study included patients ages 45 to 53 who were from the University Hospital Frankfurt Covid-19 Registry in Germany. They were recruited for the study between April and June. Most of the patients -- 67-- recovered at home, with the severity of their illness ranging from some being asymptomatic to having moderate symptoms.
The researchers used cardiac magnetic resonance imaging, blood tests and biopsy of heart tissue. Those data were compared with a group of 50 healthy volunteers and 57 volunteers with some underlying health conditions or risk factors.
The MRI data revealed that people infected with coronavirus had some sort of heart involvement regardless of any preexisting conditions, the severity or course of their infection, the time from their original diagnosis or the presence of any specific heart-related symptoms.
The most common heart-related abnormality in the Covid-19 patients was myocardial inflammation or abnormal inflammation of the heart muscle, which can weaken it.
This type of inflammation, also called myocarditis, is usually caused by a viral infection, Goldberg said, adding that she was not surprised by these study results.
"What they're saying in this study is that you can identify myocardial involvement or heart involvement by magnetic resonance imaging," Goldberg said.
The study has some limitations. More research is needed to determine whether similar findings would emerge among a larger group of patients, those younger than 18 and those currently battling coronavirus infection instead of just recovering from it.
"These findings indicate the need for ongoing investigation of the long-term cardiovascular consequences of COVID-19," the researchers wrote.
'This infection does not follow one path'
In the other JAMA Cardiology study, an analysis of autopsies found that coronavirus could be identified in the heart tissue of Covid-19 patients who died.
The study included data from 39 autopsy cases from Germany between April 8 and April 18. The patients, ages 78 to 89, had tested positive for Covid-19 and the researchers analyzed heart tissue from their autopsies.
The researchers found that 16 of the patients had virus in their heart tissue, but did not show signs of unusual sudden inflammation in the heart or myocarditis. It's not clear what this means, the researchers said.
The sample of autopsy cases was small and the "elderly age of the patients might have influenced the results," the researchers wrote. More research is needed whether similar findings would emerge among a younger group of patients.
"I think both of these studies are important," Goldberg said.
"One pretty much shows that the MRI scan can help diagnose the myocardial injury that occurs due to Covid and it was confirmed on biopsy," she said. "The autopsy study showed us something else that's interesting -- that you can have viral presence but not the acute inflammatory process. So this infection does not follow one path."
'An increasingly complex puzzle'
Both studies "add to an increasingly complex puzzle" when it comes to the novel coronavirus named SARS-CoV-2, Dr. Dave Montgomery, founding cardiologist at the PREvent Clinic in Sandy Springs, Georgia, said in an email on Tuesday.
"Taken together the studies support that SARS-CoV-2 does not have to cause clinical myocarditis in order to find the virus in large numbers and the inflammatory response in myocardial tissue. In other words, one can have no or mild symptoms of heart involvement in order to actually cause damage," said Montgomery, who was not involved in the studies.
"Viruses in general have a way of making their way to organs that are quite remote from the original site of infection. SARS-CoV-2 is no different in this regard," he said. "What is different is that this virus seems to preferentially affect cardiac cells and the surrounding cells. These studies suggest that the heart can be infected with no clear signs. Personally, in my practice, we have seen similar signs of inflammation, including pericardial effusions," or fluid around the sac of the heart.
Get CNN Health's weekly newsletter
Sign up here to get The Results Are In with Dr. Sanjay Gupta every Tuesday from the CNN Health team.
Dr. Clyde Yancy of Northwestern University Feinberg School of Medicine and Dr. Gregg Fonarow of the University of California, Los Angeles, co-authored an editorial that accompanied the two new studies in the journal JAMA Cardiology on Monday.
"We see the plot thickening and we are inclined to raise a new and very evident concern that cardiomyopathy and heart failure related to COVID-19 may potentially evolve as the natural history of this infection becomes clearer," Yancy and Fonarow wrote in the editorial.
"We wish not to generate additional anxiety but rather to incite other investigators to carefully examine existing and prospectively collect new data in other populations to con- firm or refute these findings," they wrote.
| ||
| Russian Intelligence Agencies Push Disinformation on Coronavirus Pandemic | ||
After the publication of this article, OneWorld.Press issued a statement saying any accusation that it worked for Russian intelligence was “categorically false.” “To the best of our knowledge,” its contributors have not been charged with crimes for cooperating with any foreign intelligence agency, the statement said.
Without evidence, OneWorld.Press claimed that the accusations about Russian intelligence’s propaganda efforts were being spread by officials who aimed to hurt President Trump’s re-election chances. “Everybody across the world knows that some members of the ‘deep state’ have their daggers out for Trump, and the president himself has even said as much on countless occasions,” it said.
American intelligence officials said the G.R.U.’s psychological warfare unit, known as Unit 54777 or the 72nd Special Service Center, was behind the propaganda campaigns that were often devised to obscure Moscow’s role in creating them. A 2018 report in The Washington Post linked InfoRos to the G.R.U.’s Unit 54777.
United States intelligence reports have identified two Russians, Denis V. Tyurin and Aleksandr G. Starunskiy, with ties to the G.R.U. and who make sure the messaging and disinformation drafted by the intelligence officials are pushed by InfoRos and on InfoBrics.org and OneWorld.Press.
Russian officials did not immediately return a request for comment.
Mr. Tyurin and Mr. Starunskiy, the American officials said, were in essence involved in a kind of information laundering, akin to money laundering. They take the messages from Russian intelligence and spread them on InfoRos, OnePress or another website.
The Coronavirus Outbreak ›FREQUENTLY ASKED QUESTIONS
Updated July 27, 2020
The material created by the G.R.U. is then picked up by other websites that further spread it. Those websites are often on the fringes of the web, while some, like Global Research, have a significant following, American officials said.
The stories pushed by Russian intelligence appear to be written by native English speakers and do not stand out as products of a foreign influence campaign, American officials said.
| ||
| Dynamic Changes of Antibodies to SARS-CoV-2 in COVID-19 Patients at Early Stage of Outbreak - Docwire News | ||
This article was originally published here
Virol Sin. 2020 Jul 27. doi: 10.1007/s12250-020-00268-5. Online ahead of print.
ABSTRACT
The coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2, has spread around the world with high mortality. To diagnose promptly and accurately is the vital step to effectively control its pandemic. Dynamic characteristics of SARS-CoV-2-specific antibodies which are important for diagnosis of infection have not been fully demonstrated. In this retrospective, single-center, observational study, we enrolled the initial 131 confirmed cases of COVID-19 at Jin-Yin-Tan Hospital who had at least one-time antibody tested during their hospitalization. The dynamic changes of IgM and IgG antibodies to SARS-CoV-2 nucleocapsid protein in 226 serum samples were detected by ELISA. The sensitivities of IgM and IgG ELISA detection were analyzed. Result showed that the sensitivity of the IgG ELISA detection (92.5%) was significantly higher than that of the IgM (70.8%) (P < 0.001). The meantimes of seroconversion for IgM and IgG were 6 days and 3 days, respectively. The IgM and IgG antibody levels peaked at around 18 days and 23 days, and then IgM fell to below the baseline level at about day 36, whereas IgG maintained at a relatively high level. In conclusion, antibodies should be detected to aid in diagnosis of COVID-19 infection. IgG could be a sensitive indicator for retrospective diagnosis and contact tracing, while IgM could be an indicator of early infection.
PMID:32720214 | DOI:10.1007/s12250-020-00268-5
| ||
| 9:22 AM 7/29/2020 - Russia spreads disinformation and incites the racial wars in America - their old trick | Russian intelligence spreading virus disinformation 29/07/20 08:40 from Saved Stories from Michael_Novakhov | ||
 Russia spreads disinformation and incites the racial wars in America - their old trick - GS __________________________________________________________________________ 9:22 AM 7/29/2020 - Russian intelligence spreading virus disinformation 29/07/20 08:40 from Saved Stories from Michael_Novakhov 
_____________________________________
» Saved Stories - None: 8:31 AM 7/29/2020 - Corona virus and US Military: US officials say Russian intelligence spreading virus disinformation
29/07/20 08:40 from Saved Stories from Michael_Novakhov (1 sites) By Michael Novakhov (Mike Nova) July 29, 2020 So much for Antifa - Mystery 'Umbrella Man' Vandal From Minnesota: Police Say He’s A White Supremacist Instigator ___________________________________________________________ 8:31 AM 7/2...
» Saved Stories - None: Covid-19: strains, infectivity and severityimages.app.goo.gl/JtZqGtFzeRogtY…
29/07/20 08:24 from Saved Stories from Michael_Novakhov (1 sites) Covid-19: strains, infectivity and severity images.app.goo.gl/JtZqGtFzeRogtY… Posted by mikenov on Tuesday, July 28th, 2020 8:00pm Saved Stories - None
» Saved Stories - None: Google Alert - Coronavirus and US Navy: Coronavirus COVID-19 'superspreaders' are emerging as the lepers of the 21st century
29/07/20 08:23 from Saved Stories from Michael_Novakhov (1 sites) ... nearly three decades in isolation in the US after she was blamed for several ... In many cases they have been the villains of the COVID -19 pandemic. ... In the Sri Lankan case, questions have been raised over whether the navy ...
» Saved Stories - None: Google Alert - Coronavirus and white supremacy: Politics at the point of a gun
29/07/20 08:23 from Saved Stories from Michael_Novakhov (1 sites) ... coronavirus restrictions, and rushed to the battlefield in Gettysburg following ... Groups with openly white nationalist and neo-Nazi views also have ... supported white nationalism, white separatism or white supremacy in some form. ...
» Saved Stories - None: Google Alert - Coronavirus and failure of US Intelligence Services: Global Order in the Shadow of the Coronavirus: China, Russia and the West
29/07/20 08:22 from Saved Stories from Michael_Novakhov (1 sites) The crisis of the liberal order reflects a collective Western failure to live up to its ... They are central to the Trump Administration's openly combative approach ... The latest iterations of the US National Security Strategy (2017) an...
» Saved Stories - None: Google Alert - sars cov 2: Dynamic Changes of Antibodies to SARS-CoV-2 in COVID-19 Patients at Early Stage of Outbreak
29/07/20 08:22 from Saved Stories from Michael_Novakhov (1 sites) Online ahead of print. ABSTRACT. The coronavirus disease 2019 (COVID-19), caused by SARS - CoV - 2 , has spread around ... Google Alert - sars cov 2 Saved Stories - None
» Saved Stories - None: Google Alert - coronavirus in new york post: Cuomo blames de Blasio's Rikers releases, anti-cop attitude for NYC crime wave
29/07/20 08:21 from Saved Stories from Michael_Novakhov (1 sites) “In terms of overall violence in New York City: I understand what the mayor said about the ... “A number of people were let out of Rikers during COVID . Google Alert - coronavirus in new york post Saved Stories - None
» Saved Stories - None: Google Alert – coronavirus and orthodox jews: In Western Europe, a Jewish ‘Community’ is an organization. They operate like expensive …
29/07/20 08:21 from Saved Stories from Michael_Novakhov (1 sites) … of French Jews overcome the financial repercussions of the coronavirus crisis. … In Poland, a 2012 court ruling forced the Orthodox Jewish Community of … When the non- Orthodox congregation joined the Community, some ...
» Saved Stories - None: Google Alert - Coronavirus and US Navy: US Navy is studying 3500 teenage Marine recruits in an effort to learn why coronavirus makes ...
29/07/20 08:21 from Saved Stories from Michael_Novakhov (1 sites) The US Navy is looking to recruit 3,500 18- and 19-year-old Marine recruits for its COVID -19 Health Action Response for Marines study; Recruits ... Google Alert - Coronavirus and US Navy Saved Stories - None
» Saved Stories - None: Google Alert - coronavirus in new york post: Misleading Virus Video, Pushed by the Trumps, Spreads Online
29/07/20 08:20 from Saved Stories from Michael_Novakhov (1 sites) In a video posted Monday online, a group of people calling themselves ... coronavirus treatment and that masks did not slow the spread of the virus. ... At least one version of the video, viewed by The Times on Facebook, was ... Clockwis... | ||
| Russia spreads disinformation and incites the racial wars in America - their old trick - Google Search | ||
Search ResultsWeb resultsHow Russia Tried to Start a Race War in the United States
repository.law.umich.edu › cgi › viewcontent
repository.law.umich.edu › cgi › viewcontent
PDF
This. Article examines Russia's actions and considers whether they violate the international prohibitions against racial discrimination and hate speech. Table of
by WJ Aceves - 2019 - Cited by 2 - Related articles
Russia Trying to Stoke U.S. Racial Tensions Before Election ...
www.nytimes.com › U.S. › Politics
www.nytimes.com › U.S. › Politics
Mar 10, 2020 - Russian intelligence services are trying to incite violence by white supremacist groups to sow chaos in the United States, American intelligence ...
Russian Intelligence Agencies Push Disinformation on ...
www.nytimes.com › U.S. › Politics
www.nytimes.com › U.S. › Politics
16 hours ago - Declassified U.S. intelligence accuses Moscow of pushing propaganda through alternative websites as Russia refines techniques used in ...
Missing:
The History of Russian Involvement in America's Race Wars ...
www.theatlantic.com › 2017/10 › russia-facebook-race
www.theatlantic.com › 2017/10 › russia-facebook-race
Oct 21, 2017 - From propaganda posters to Facebook ads, 80-plus years of Russian meddling.
|
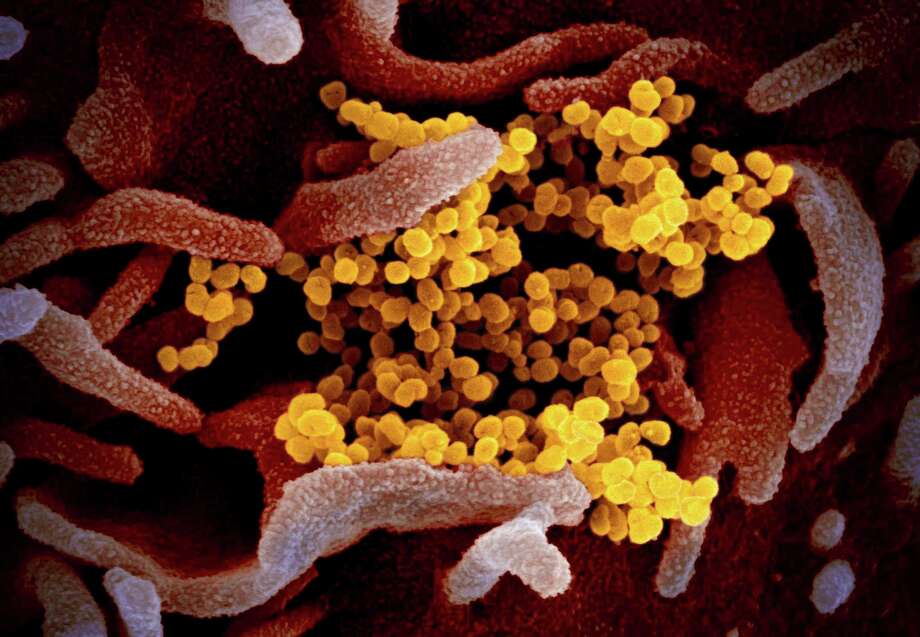
























Trump and Trumpism – Review Of News And Opinions: 7:53 AM 7/2/2019 – Michael Novakhov – SharedNewsLinks℠: 6:58 AM 7/2/2019 – “Fear, and the paralysis…”: Trump corruption investigators risk jobs, reputations for seeking truth – By Michael J. Stern | Trump and Trumpism – Review Of News And Opinions – Topic Review: New Abwehr, WW2, Japan, Korea
_______________________________________
Douglas Leff: Brief Psychological Portrait as the impressions from the photograph. – By Michael Novakhov – 10:18 AM 8/6/2019
Did Douglas Leff Manufacture The Case Of Julia Keleher, and why? – Google Search | ||
Douglas Leff: Brief Psychological Portrait as the impressions from the photograph. – By Michael Novakhov – 10:18 AM 8/6/2019 – Post Link
 This guy looks too complex, too smart, too ambitious, and too psychopathic – for the price of his suit, whatever meaning you want to put into it. It means that he is masking, plays (but all of them are bad actors, and it shows), lies, hides (his wealth, first of all), is perverse; loves money and power above all. He also appears to be very controlling, deeply paranoid (suspicious about everything), apprehensive, defensive.But overall, he is hard to read. As I posted earlier, he fits the concept of the proverbial onion: many masking and protective layers but no substance and no essence inside, except for money and power, his psychological drivers.What you, or rather I, see in the depth of his eyes, is THE BEAST. The same naked, fearless, merciless, low, playful, mysterious, animalistic, Hebrew-Abwehr mix, Mafia Beast, that you can see in many of the faces of their leaders (and he would be in the first row, definitely) but extremely and uniquely well masked. In my humble opinion.
M.N.
10:17 AM 8/6/2019
________________________________________________________
|







Comments
Post a Comment